
Общие сведения
Невральная амиотрофия Шарко Мари Тута объединяет группу наследственных прогрессирующих хронически протекающих полиневропатий:
- болезнь Рефсума;
- гипертрофическая невропатия Дежерина-Сотта;
- синдром Русси-Леви.
Частота встречаемости варьирует от 2 до 36 случаев на 100 тысяч населения. Невральная амиотрофия носит в основном семейный характер и у разных родственников клиническая симптоматика может сильно разниться. Параллельно регистрируются спорадические варианты течения болезни Шарко-Мари-Тута. Невральная амиотрофия чаще поражает лиц мужского пола.
Болезнь Шарко Мари Тута относится сразу к нескольким заболеваниям, которые получили название от имени Жана-Мартина Шарко:
- Болезнь Лу Герига или боковой амиотрофический синдром– дегенеративное мышечное заболевание. По данным «Википедия» заболевание получило название от имени знаменитого американского бейсболиста Генри Луи Герига, страдавшего данной патологией. Еще одно название – боковой амиотрофический склероз.
- Артропатия или болезнь Шарко – нейропатическая артропатия. Характеризуется прогрессирующей дегенерацией весового сустава.
- Синдром Шарко Мари Тута – демиелинизирующее наследственное заболевание, поражающие периферическую нервную систему (источник Википедия).
Патогенез
Большинство форм болезни связано с поражением миелиновых оболочек в волокнах периферических нервов. Гораздо реже встречается патология аксонов (осевые цилиндры, которые проходят по центру нервного волокна). Дегенеративные изменения наблюдаются также в путях Голля (проводящая система глубокой чувствительности в спинном мозге), нейронах передних рогов, корешках спинного мозга (передние и задние), столбах Кларка (относятся к заднему спинномозжечковому пути).
На фоне дисфункции периферической нервной системы развивается мышечная атрофия, которая затрагивает отдельные группы миофибрилл. По мере прогрессирования патологии происходит смещение ядер сарколеммы, интерстициальное разрастание соединительной ткани, поражение миофибрилл. При нарастании гиалиновой дегенерации миофибрилл наблюдается их распад.
Классификация
Выделяют 2 типа невральной амиотрофии Шарко. Разграничение основывается на ряде особенностей, но в целом клиническая симптоматика у обоих типов схожа.
- I тип. Характерно выраженное снижение скорости проведения нервных импульсов. При биопсии нерва выявляется сегментарная демиелинизация нервных волокон и гипертрофическое разрастание непоражённых шванновских клеток.
- II тип. Скорость проведения импульсов практически не страдает, а при анализе биоптата выявляется дегенерация аксонов.
Также есть связь между атаксией Фридрейха и болезнью Шарко-Мари-Тута. У некоторых пациентов с ШМТ диагностируется клиническая симптоматика атаксии Фридрейха и наоборот (в течением времени). Также встречаются промежуточные формы этих заболеваний. В медицинской практике описаны случаи выявления атаксии Фридрейха у одних родственников и амиотрофии ШМТ – у других.
Причины
Достоверных данных о причинах развития невральной амиотрофии на сегодняшний день в практической неврологии нет. У 70-80% пациентов с болезнью Шарко отмечалось дублирование 1 участка 17 хромосомы. У патологии есть несколько форм, что обусловлено мутациями различных генов. Для заболевания характерен аутосомно-доминантный тип наследования с показателем пенетрантности на уровне 83%. Также были зарегистрированы случаи аутосомно-рецессивного типа наследования.
Симптомы болезни Шарко
Заболевание начинается с симметричной атрофии мышц в дистальных отделах нижних конечностей. Первая симптоматика появляется в 10-15 лет, гораздо реже в 16-30. Пациенты отмечают быструю утомляемость в стопах при длительном стоянии на одном месте. Регистрируется также симптом «топтания» — пациент перетаптывается на одном месте, чтобы снять нагрузку.
В некоторых случаях заболевание начинается с нарушения чувствительности в стопах, и очень часто – с парестезий (чувство ползания мурашек по стопам). Характерным ранним признаком заболевания является отсутствие сначала ахилловых, а затем и сухожильных, коленных рефлексов. Часто пациенты предъявляют жалобы на болезненные, приступообразные сокращения в икроножных мышцах, которые усиливаются после длительной физической активности либо в ночное время.
Первоначально атрофия затрагивает только разгибатели и абдукторы стопы, что приводит к свисанию стопы и невозможности ходить на пятках, появляется характерная, специфическая походка, похожая на степпаж – вышагивание лошади. Далее атрофия распространяется на мышечную систему и сгибатели стопы. При тотальном поражении мышц стопы развивается её деформация с высоким сводом аналогично стопе Фридрейха, пальцы приобретают специфическую молоткообразную форму. По мере прогрессирования патологический процесс затрагивает проксимальные отделы ног – нижнюю часть бёдер и голени, развивается «болтающаяся стопа», а ноги приобретают форму перевёрнутых бутылок.
Далее болезнь прогрессирует и поражает дистальные отделы рук – сначала кисти, а затем атрофия касается и мышц предплечий. Кисть становится похожей на лапу обезьяны из-за поражения тенара и гипотенара. Атрофические изменения никогда не регистрируются в мышцах плечевого пояса, туловища и шеи.
Очень часто отмечаются лёгкие фасцикулярные подёргивания мышц рук и ног. В некоторых случаях появляется компенсаторная гипертрофия в мышцах проксимальных отделов конечностей. Чувствительные нарушения проявляются тотальной гиперстезией. Болевая и температурная чувствительность страдают больше, чем глубокая. Редко появляется отёчность в поражённых конечностях и цианоз кожных покровов.
Клинические симптомы при болезни ШМТ прогрессируют очень медленно. Временной промежуток между поражением верхнего и нижнего пояса может достигать 10 лет. Длительное время пациенты остаются трудоспособными. Негативное влияние на скорость развития патологического процесса могут оказывать разные экзогенные факторы:
- черепно-мозговая травма;
- переохлаждение;
- перенесённые инфекции (краснуха, мононуклеоз, корь, ОРВИ, ангина);
- гиповитаминоз;
- травма позвоночника, спинного мозга.
Анализы и диагностика
Обследованием пациентов с подозрением на Болезнь Шарко-Мари-Тута занимаются невропатологи и ортопеды. При первичном осмотре уточняется возраст пациента, в котором впервые стала появляться характерная симптоматика. Обязателен сбор семейного анамнеза, с уточнением наличия у родственников схожих генетических заболеваний. При осмотре врач обращает внимание на деформацию кистей рук и стоп ног, на изменение походки.
Во время неврологического осмотра выявляется снижение тонуса в дистальных отделах верхних и нижних конечностей, снижение чувствительности кожных покровов и ослабление (вплоть до полного отсутствия) сухожильных рефлексов (коленные, ахилловы).
Основные методы исследования:
- ЭНМГ. Диагностируются признаки демиелинизирующей и аксональной нейропатии, что проявляется падением амплитуды М-ответов, снижением скорости проведения импульсов по двигательным нервам.
- Гистология. При анализе биоптата из большеберцового нерва отмечается атрофия миелина, снижение количества миелиновых волокон, разрастание соединительно-тканных волокон. При проведении биопсии нервного волокна выявляется характерный морфологический признак – «луковичные головки».
- Компьютерная паллестезиометрия. Диагностическая процедура позволяет выявить наличие или отсутствие снижения вибрационной чувствительности, которое является самым ранним признаком проявления заболевания.
- ДНК-анализ. Для полной верификации диагноза проводится анализ ДНК, который позволяет выявить дупликацию гена РМР22 – белка периферического миелина на 17 хромосоме.
Дифференциальная диагностика проводится с заболеваниями:
- синдром Гийена-Барре;
- адренолейкодистрофия;
- спинальная мышечная атрофия Верднига-Гоффмана;
- болезнь Пелицеуса-Мерцбахера.
Лечение
Терапия пациентов с болезнью Шато проводится в стационарных условиях. Какого-либо специфического лечения нет, чтобы замедлить прогрессирование демиелинизации и аксональной дегенерации. Индивидуальная, грамотно подобранная терапия позволяет улучшить качество жизни пациента.
Доктора
Лекарства
Медикаменты, которые применяются при невральной амиотрофии ШМТ:
- Витамины. Витаминные комплексы позволяют восстановить структуру нервных волокон, улучшить микроциркуляцию. С особой осторожностью назначают витамин В6 из-за его нейротоксичности. Проведённые исследования показали, что аскорбиновая кислота способна подавлять синтез РМР22 периферического белка миелина.
- Витамин Д и кальций. У 40% пациентов диагностируется остеопороз, что требует назначения Колекальциферола и препаратов кальция для профилактики переломов.
- Миорелаксанты. Для уменьшения выраженности болевого синдрома от мышечных сокращений назначают препараты, которые расслабляют скелетную мускулатуру (Толперизон, Баклофен).
- Антихолинэстеразные препараты. Для улучшения нервно-мышечной проводимости при ШМТ 2 типа назначают Галантамин, Прозерин.
Процедуры и операции
Особое внимание уделяется немедикаментозным методам терапии. Комплекс мероприятий, позволяющих достичь максимального терапевтического эффекта:
- Лечебная физическая культура. Регулярные занятия ЛФК повышают мышечный тонус. Наибольший эффект достигается при достижении пассивных упражнений (занятия вместе со специалистом) и активных (выполняются самостоятельно).
- Электростимуляция. Направленная подача электрических импульсов улучшает проводимость по периферической нервной системе, активирует метаболические процессы в паретичных мышцах, усиливает нейротрофику.
- Бальнеотерапия. Грязевые аппликации и грязевые ванны замедляют формирование контрактур и улучшают работу вегетативной нервной системы.
- Массаж. Различные виды массажа (аппаратный и ручной) улучшают лимфатический отток и нормализуют кровообращение. Рекомендован расслабляющий, стимулирующий и вибрационный массаж.
- Ортопедическая терапия. Ношение специальной ортопедической обуви позволяет предотвратить развитие грубой деформации. Из-за мышечной слабости развивается нестабильность суставов. Подтяжки, ортезы и другие специальные приспособления используются для фиксации стоп в необходимом положении.
При комплексном подходе к терапии повышается уровень мышечной силы, нормализуется походка и исправляются различные нарушения равновесия. Грамотно подобранное лечение повышает социальную и бытовую адаптацию, восстанавливает работоспособность пациента.
Хирургическое лечение
При выраженной деформации стопы и развитии атрофических изменений, которые затрудняют самостоятельную ходьбу, а также при отсутствии эффекта от консервативной терапии, показано специальное ортопедическое оперативное вмешательство:
- остеотомия пяточной кости;
- метатарзальная остеотомия.
Артродез проводится с целью восстановления опорной функции стопы.
Прогноз и профилактика
Невральная амиотрофия Шарко Мари считается тяжёлым инвалидизирующим заболеванием. Пациенты теряют способность к самостоятельной ходьбе через 15-20 лет от дебюта заболевания. Продолжительность жизни пациентов практически не отличается от общей популяции, что объясняется поражением преимущественно дистальных отделов конечностей.
При сочетании патологии с атаксией Фридрейха в молодом возрасте очень высока вероятность летального исхода, что объясняется вовлечением в патологический процесс миокарда и дыхательной мускулатуры.
Специфического метода первичной профилактики нет. Своевременно начатая грамотная терапия позволяет избежать осложнений и сохранить работоспособность на максимально продолжительный период.
Список источников
- Дадали Е.Л., Тибуркова Т.Б., Щагина О.А., Поляков А.В. «Алгоритм диагностики наследственных моторно-сенсорных невропатий», статья из журнала «Нервные болезни», 2010
- Муртазина А.Ф., Щагина О.А, Никитин С.С., Дадали Е.Л. , Поляков А.В. «Современные клинико-генетические представления об аутосомно-рецессивных наследственных периферических нейропатиях», статья в журнале «Анналы клинической и экспериментальной неврологии» №1, 2019
- Каук О.И. «Наследственные полиневропатии как фактор статокинетических нарушений у детей раннего возраста» статья в Международном медицинском журнале №1, 2016
Источник: medside.ru
Описание заболевания
Синдром Шарко (или перемежающаяся хромота) представляет собой заболевание, которое характеризуется возникновением, а также усилением болевых ощущений и слабости в нижних конечностях во время ходьбы. Такие проявления часто заставляют больного останавливаться, ведь в покое названные симптомы практически не беспокоят. Чаще всего этому отклонению люди подвергаются в результате:
- чрезмерного увлечения алкогольными напитками и табаком;
- лишнего веса;
- повышенного уровня холестерина в крови;
- наследственности и проч.
Основные симптомы
Синдром Шарко – это довольно болезненное состояние, которое причиняет больному неудобство и значительно ухудшает качество его жизни.
и таком отклонении человек часто жалуется на чувство усталости, а также дискомфорт и боли в нижних конечностях во время ходьбы, особенно в ягодичной области и икроножных мышцах. Бывают и такие случаи, когда болевые ощущения локализуются в мышечных тканях бедер и пояснице. Как отмечают пациенты с диагнозом Шарко, болезнь частично затихает и ослабевает сразу же после непродолжительного отдыха.
При таком заболевании в дистальных отделах нижних конечностей у больного часто наблюдаются трофические и вегетативно-сосудистые нарушения (например, акроцианоз, похолодание стоп, мраморность кожных покровов и их дистрофические изменения, отсутствие пульса на артериях стоп, а также гангрена пальцев, которая часто распространяется в проксимальном направлении).
Помимо всего прочего, синдром Шарко характеризуется и дегенеративно-дистрофическими изменениями костей и суставов (гипертрофия отдельных участков, дегенерация хрящей, костные секвестры, остеофиты, внутрисуставные и спонтанные переломы трубчатых костей). Кроме того, разболтанность и пониженная чувствительность суставов значительно увеличивают риск появления травматических повреждений.
Причины возникновения
Синдром Шарко – болезнь, обусловленная поражением артерий ног (тромбангиитом, облитерирующим атеросклерозом, периферической формой неспецифического аортоартериита и проч.). Болевые ощущения у пациента возникают из-за недостаточного притока крови к мышечным тканям нижних конечностей. Также заболевание может возникнуть в результате сдавления конского хвоста при стенозе позвоночника, реже – вследствие ишемии спинного мозга при артериовенозной мальформации или атеросклерозе аорты.
Лечение заболевания
Терапия перемежающейся хромоты заключается в непосредственном лечении основного источника заболевания, а именно артерий. Как правило, в таких ситуациях врачи советуют своим пациентам отказаться от всех вредных привычек, снизить вес и соблюдать строгую диету. Также пациентам назначается специальный комплекс физических упражнений, которые позволят победить признаки болезни.
Что касается консервативных методов лечения, то к ним можно отнести прием различных медицинских препаратов. Их действие дает обезболивающий и сосудорасширяющий эффект. Также аптечные средства способствуют понижению холестерина в крови и ее разжижению.
При острой необходимости применяется хирургическое вмешательство, в процессе которого расширяется просвет артерии при помощи введения в нее специального катетера.
Синдром Шарко-Мари-Тута
Данное отклонение представляет собой группу наследственных заболеваний, для которых характерна дегенерация периферических нервных волокон. Следует особо отметить, что разные варианты названного синдрома могут основываться на разных генетических дефектах. Большинство таких болезней наследуются по аутосомно-доминантному механизму. Однако в настоящее время выявлены и варианты заболеваний, связанные со сцеплением Х-хромосомы.
При таком диагнозе, как синдром Шарко-Мари, у больного поражаются спинной мозг и периферические нервы. В зависимости от определенного генетического дефекта у человека может формироваться аксональная или демиелинизирующая невропатия.
Чаще всего представленное отклонение начинает развиваться в юношеском или подростковом возрасте. Симптомы этой группы заболевания со временем усиливаются, а болезнь прогрессирует, в результате чего наступает умеренная степень инвалидизации, не приводящая к летальному исходу.
Симптомы отклонения
Данная болезнь начинает проявляться с атрофии дистальных мышечных тканей ног и слабости. Со временем стопы деформируются, а пальцы становятся молоткообразными и большими. У детей с таким диагнозом практически всегда наблюдается задержка физического развития.
Лечение наследственной болезни
Определенной терапии такого патологического состояния не существует. Однако для улучшения самочувствия больного врачи часто назначают лечебную гимнастику и трудотерапию. Кроме того, пациентами с таким диагнозом активно используются всевозможные медицинские приспособления, позволяющие устранить болевые симптомы. К примеру, при висячей стопе применяют ортопедическую скобу, укрепляющую голень и проч. Также имеет довольно большое значение генетическое консультирование.
Болезнь фон Виллебранда
Следует особо отметить, что к наследственным заболеваниям относится не только синдром Шарко. Виллебранда болезнь также может передаваться от ближайших родственников.
Как известно, представленное отклонение является наиболее распространенным видом наследственных расстройств, приводящих к нарушению свертывания крови или процессу коагуляции. Однако нередко это заболевание возникает и в течение жизни в результате действия других болезней (то есть приобретенная форма).
Симптомы болезни
Риск возникновения кровотечения при таком отклонении значительно отличается в зависимости от того или иного типа болезни. Основными признаками этого наследственного расстройства являются периодические кровотечения из носа, беспричинное появление гематом и синяков, кровоточивость десен и проч. У женщин могут становиться обильными месячные, а при родах существует риск потери большого количества крови.
Лечение заболевания
Пациентам с таким диагнозом никакого лечения не требуется, но риск обильного кровотечения у них всегда повышен. Таким образом, больному могут быть рекомендованы препараты, которые снижают этот явный симптом заболевания. Если же пациенту планируется делать хирургическую операцию, то врачи обязательно проводят профилактическое лечение.
Источник: FB.ru
| Процедуры и операции | Средняя цена |
| Гастроэнтерология / Диагностика в гастроэнтерологии / УЗИ в гастроэнтерологии | от 170 р. 1122 адреса |
| Стоматология / Консультации в стоматологии | от 100 р. 890 адресов |
| Отоларингология / Консультации в отоларингологии | от 563 р. 726 адресов |
| Гастроэнтерология / Консультации в гастроэнтерологии | от 563 р. 706 адресов |
| Гинекология / Операции на матке / Удаление матки | 85142 р. 163 адреса |
| Онкология / Консультации в онкологии и гематологии | от 800 р. 153 адреса |
| Педиатрия / Консультации детских специалистов / Первичные консультации детских специалистов | от 1500 р. 139 адресов |
| Гинекология / Операции на матке / Удаление матки | 174107 р. 74 адреса |
| Онкология / Консультации в онкологии и гематологии | от 600 р. 56 адресов |
| Хирургия / Анестезия и реанимация / Гемотрансфузии | 7400 р. 52 адреса |
Источник: www.KrasotaiMedicina.ru
Эпидемиология[править | править код]
Распространенность болезни Виллебранда составляет 1 на 800—1000. В отдельных источниках указывается, что частота случаев равна 2%[3].
Этиология[править | править код]
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда, который участвует в адгезии тромбоцитов на коллагене и защищает VIII фактор от протеолиза. При дефиците Фактора Виллебранда VIII фактор подвергается протеолизу, и его содержание в плазме снижается. Кроме того, при болезни Виллебранда снижается содержание серотонина и развивается патологическая дилатация сосудов и повышение их проницаемости. При болезни Виллебранда наблюдаются самые длинные кровотечения, т.к. у больных нарушены все три звена гемостаза.
Синонимы[править | править код]
- Ангиогемофилия;
- Атромбопеническая пурпура;
- Атромбоцитопеническая пурпура;
- Геморрагическая капилляропатия;
- Конституциональная тромбопатия, конституциональная тромбопатия фон Виллебранда-Юргенса;
- Наследственная псевдогемофилия; синдром Юргенса;
- Сосудистая гемофилия, псевдогемофилия.
Классификация[править | править код]
Различают три типа болезни Виллебранда.
- 1-й тип обусловлен частичным количественным дефицитом фактора Виллебранда. При этом мультимерная структура его сохранена. Имеется снижение прокоагулянтной активности фактора VIII, агрегации тромбоцитов, индуцированной ристоцетином, ристоцетинкофакторной активности, антигена фактора Виллебранда. Частота данной формы составляет от 75 % до 80 % всех случаев болезни Виллебранда. Наследование аутосомно-доминантное.
- 2-й тип обусловлен качественными изменениями фактора Виллебранда, связанными с нарушением формирования мультимеров, и подразделяется на подтипы: 2A, 2B, 2M, 2N.
-
- Фенотип подтипа 2A является результатом нарушения двух различных механизмов: дефекта синтеза высокомолекулярных мультимеров и повышения протеолиза фактора Виллебранда. При подтипе 2B отмечается повышенное сродство фактора Виллебранда к рецептору на мембране тромбоцитов гликопротеину Ib.
-
- Подтип 2M характеризуется нарушением связи фактора Виллебранда с рецептором гликопротеином Ib на мембране тромбоцитов.
-
- Подтип 2N характеризуется нормальным уровнем фактора Виллебранда и низкой прокоагулянтной активностью, что обусловлено нарушением связи фактора VIII и фактора Виллебранда.
Наследование болезни Виллебранда 2-го типа аутосомно-доминантное, за исключением подтипа 2N, где оно рецессивное. Частота встречаемости данных форм составляет от 5 % до 15 % всех случаев болезни Виллебранда.
- 3-й тип — наиболее тяжелая форма с полным дефицитом фактора Виллебранда. Эта форма характеризуется отсутствием фактора Виллебранда в плазме, тромбоцитах и сосудистой стенке. Уровень фактора VIII ниже 10 %. Наследование — аутосомно-рецессивное. Заболевание проявляется у гомозигот с одинаковыми дефектными аллелями или двойных гетерозигот с двумя различными дефектными аллелями. У пациентов с 3-м типом имеется вероятность появления аллоантител к фактору Виллебранда. Частота встречаемости заболевания 3-го типа болезни Виллебранда менее 5 %.
Кроме того, существует тромбоцитарный тип болезни Виллебранда, который обусловлен мутацией в гене тромбоцитарного рецептора гликопротеина Ib, вследствие которой повышается чувствительность данного рецептора к высокомолекулярным мультимерам фактора Виллебранда. Фенотип аналогичен подтипу 2B.
- Приобретенный синдром Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями, обусловлен появлением ингибитора против фактора Виллебранда, а также качественными аномалиями фактора VIII в связи с адсорбцией высокомолекулярных мультимеров патологическими белками.
Клиническая картина[править | править код]
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из слизистых полости рта, носа, внутренних органов. Симптомы кровоточивости варьируют от умеренно выраженных до крайне тяжелых, протекают преимущественно по микроциркуляторному типу. У пациентов с резким дефицитом фактора VIII наблюдаются обильные и продолжительные кровотечения (носовые, десневые, маточные), также кровоизлияния в мышцы и суставы. Кроме того, могут возникать длительные кровотечения при травмах, удалении зубов, операциях.
В детском возрасте часто бывают кровотечения из слизистых оболочек полости рта, носовые кровотечения, синяки на коже. Более тяжелое течение геморрагического диатеза отмечается во время или вскоре после перенесенных инфекционных заболеваний. Наиболее вероятным пусковым механизмом кровотечения на фоне инфекции является нарушение проницаемости сосудов. Вследствие этого появляются самопроизвольные кровотечения диапедезного типа.
Гематомы — кровоизлияния в подкожную клетчатку и мышечные ткани наблюдаются преимущественно после травм у больных с тяжелыми формами заболевания.
При болезни Виллебранда геморрагический синдром проявляется не всегда, периоды обострения чередуются с периодами полного или почти полного отсутствия геморрагий. У некоторых пациентов болезнь Виллебранда может сочетаться с признаками мезенхимальной дисплазии: повышенной растяжимостью кожи, слабостью связок с повышенной подвижностью суставов, пролабированием створок клапанов сердца.
Аутосомный тип наследования обусловливает одинаковую частоту возникновения болезни Виллебранда у пациентов обоих полов. У женщин вследствие особенностей физиологического строения организма, связанных с репродуктивной функцией, наблюдается более частое проявление геморрагических симптомов. Около 65 % женщин с болезнью Виллебранда страдают меноррагиями. Рецидивирующие маточные кровотечения, продолжающиеся более 10 дней, сопровождаются постгеморрагической анемией.
Желудочно-кишечные кровотечения у пациентов с болезнью Виллебранда не являются преобладающей формой кровоточивости. Они могут быть вызваны приемом препаратов, влияющих на агрегацию тромбоцитов (ацетилсалициловая кислота и другие нестероидные противовоспалительные средства). Кроме того, источниками кровотечений являются латентные язвы желудка и двенадцатиперстной кишки, также эрозивные гастриты, геморроидальные узлы.
У пациентов с болезнью Виллебранда могут быть длительные кровотечения при операциях, у женщин — во время родов. Роды у женщин с болезнью Виллебранда связаны с риском возникновения значительной кровопотери. У большинства пациентов со среднетяжелой и легкими формами заболевания во время беременности уровень фактора VIII повышается в 2—3 раза и достигает нормальных значений, однако в послеродовом периоде возвращается к исходному уровню.
Гемартроз — наиболее редкое проявление болезни Виллебранда, характерное для заболевания 3-го типа. Острый гемартроз сопровождается болевым синдромом, обусловленным повышением внутрисуставного давления. Сустав увеличен в объеме, кожа над ним гиперемирована и горячая на ощупь. Если гемартроз возник после травмы, нужно исключить дополнительные повреждения (внутрисуставной перелом, отрыв мыщелка, ущемление тканей). Рецидивирующие гемартрозы вызывают хронический синовит. На стадии синовита синовиальная оболочка гипертрофируется и становится основным источником кровоизлияния в сустав. При остром синовите гемартрозы могут рецидивировать, несмотря на трансфузии фактора свертывания VIII, что обусловлено воспалительным процессом в синовиальной оболочке. При хроническом синовите болевой синдром может отсутствовать, поскольку разрушена капсула сустава.
В отличие от гемофилии, при болезни Виллебранда дальнейшего прогрессирования патологического процесса и развития деформирующего остеоартроза, как правило, не наблюдается.
Кровоизлияния в головной и спинной мозг и их оболочки при болезни Виллебранда возникают в связи с травмой. В отдельных случаях причиной таких кровоизлияний может быть гипертонический криз или прием препаратов, значительно нарушающих гемостатическую функцию тромбоцитов (ацетилсалициловая кислота, бутадион и др.).
Учитывая аутосомно-доминантный тип наследования, генетический риск для потомства составляет 50 % независимо от пола плода[4].
Лечение[править | править код]
Лечение зависит от типа заболевания. Выделяют два основных способа. При первом используются препараты плазмы с высоким содержанием фактора Виллебранда или препараты фактора VIII. В легких случаях может быть достаточно одного введения, при тяжелых травмах и операциях препараты вводят дважды в сутки 2—3 дня. Второй подход к лечению применим для легких форм. Пациентам назначается десмопрессин. Существует опасность привыкания при использовании десмопрессина более 2-х суток.
Источник: ru.wikipedia.org
Что такое болезнь Виллебранда?
Болезнь Виллебранда (БВ) является распространенным наследственным нарушением свертываемости крови, затрагивающим мужчин и женщин в равной степени, но на женщин может оказать непропорционально большое влияние из-за проблем с менструацией и родами.
Существует три основных типа БВ (БВ тип 1, БФВ тип 2 и БВ тип 3), каждый с различной степенью серьезности и типами наследования. В отличие от гемофилии, которая характеризуется кровотечением в суставах, болезнь Виллебранда обычно характеризуется кожно-слизистым кровотечением. БВ вызван дефектом или дефицитом фактора фон Виллебранда (англ. von Willebrand factor, VWF), большого белка, состоящего из нескольких субъединиц.
VWF связывается с фактором свертывания VIII в кровообращении и защищает его от разрушения. VWF также помогает тромбоцитам связываться с внутренней частью поврежденных кровеносных сосудов. Это приводит к образованию стабильного сгустка крови, который закупоривает поврежденный кровеносный сосуд и останавливает кровотечение. Если VWF недостаточно, или он неисправен, у человека могут возникнуть проблемы с образованием тромба.
У большинства больных людей относительно легкая форма заболевания, БВ 1 типа, и не диагностируется до зрелого возраста. Небольшой процент этих людей может иметь длительное кровотечение в младенчестве или в раннем детстве. Симптомы могут включать кровотечение из носа, кровотечение из десен и легкие кровоподтеки. У женщин с БВ часто возникают тяжелые менструации. Пострадавшие женщины легко поддаются кровотечению после травмы, родов и/или хирургического вмешательства. Кровотечение из желудка и кишечника может возникнуть, но встречается реже.
БВ был впервые описан в медицинской литературе в 1926 году доктором Эриком Адольфом фон Виллебрандом, который дифференцировал расстройство от классической гемофилии. В дополнение к генетической форме, болезнь Виллебранда может быть приобретен в течение жизни, часто в связи с отдельным основным заболеванием.
Признаки и симптомы

Специфические симптомы и степень тяжести БВ могут сильно варьироваться от одного человека к другому, даже среди людей одного и того же подтипа или членов одной семьи. У некоторых людей могут не быть симптомов или быть только легкие проявления расстройства; у других людей могут быть от умеренных симптомов до тяжелых осложнений кровотечения. Некоторые люди могут не испытывать симптомы до взрослой жизни; другие могут быть затронуты в младенчестве.
У большинства людей легкая форма расстройства. 1 тип заболевания является преобладающей формой в общей популяции. Тяжелые симптомы чаще всего возникают при БВ типа 3 и в некоторых случаях БВ 2 типа.
Чтобы лучше понять болезнь Виллебранда, важно понять, как тело формирует сгустки (тромбы), чтобы останавливать кровотечения. Свертывание — процесс, при котором кровь собирается вместе, чтобы закрыть участок раны и остановить кровотечение. Свертывание требует ряда реакций между свертывающими белками, чтобы в конечном счете сформировать сгусток. Факторы свертывания крови, такие как VWF, представляют собой специализированные белки, которые играют важную роль в обеспечении свертываемости крови. VWF выполняет две основные функции: способствует адгезии тромбоцитов к поврежденному кровеносному сосуду и стабилизирует, защищает и переносит фактор VIII к месту повреждения. Дефекты уровней или неправильная функция VWF затрудняют процесс свертывания. Следовательно, пострадавшим людям трудно остановить кровотечение из раны.
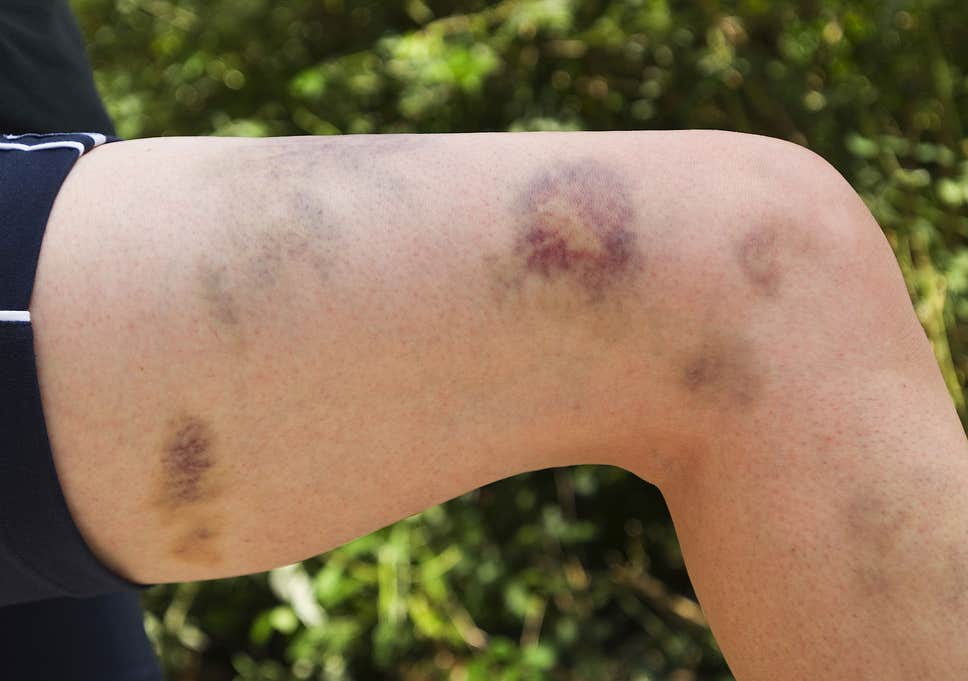
Легкие симптомы, связанные с болезнью Виллебранда, включают легкое кровотечение слизистых оболочек и кожи (слизисто-кожные участки), включая хронические носовые кровотечения и кровотечение из десен. Также могут возникнуть легкие кровоподтеки и длительное кровотечение из-за небольших порезов. На большие участки тела могут распространяться синяки. Женщины могут испытывать сильное и длительное кровотечение во время менструального цикла (меноррагия) или во время и после родов. При отсутствии лечения обильные менструальные кровотечения могут привести к анемии и дефициту железа. Некоторые люди могут испытывать тяжелое, длительное кровотечение после травмы, стоматологических процедур или хирургического вмешательства.
Более серьезные, но необычные осложнения могут включать желудочно-кишечные кровотечения, твердые отеки застывшей крови (гематомы) и кровотечения в мышцах и суставах (гемартроз), вызывающие прогрессирующее повреждение и дегенерацию суставов. В конечном итоге, в этих случаях БВ может ограничить диапазон движения пораженного сустава.
— Подтипы болезни Виллебранда.
БВ обычно разбиты на три подтипа.
- БВ 1 типа является самой легкой формой расстройства и составляет примерно 70-80% случаев. Пострадавшие люди могут иметь низкие уровни VWF в крови. В некоторых случаях фактор VIII также может быть уменьшен. Как правило, у пораженных людей развивается легкое слизисто-кожное кровотечение; в редких случаях у пострадавших людей будут развиваться более серьезные симптомы. Носовые кровотечения и кровоподтеки являются общими признаками заболевания у детей; тяжелые менструальные кровотечения — обычное явление для женщин детородного возраста.
- БВ 2 типа составляет примерно 20% случаев. У этих людей VWF может присутствовать в крови на нормальном или почти нормальном уровне, но не функционирует должным образом. БВ 2 типа дополнительно подразделяется в зависимости от конкретного, лежащего в основе дефекта, связанного с VWF. Эти подтипы известны как БВ типа 2a, 2b, 2m и 2n.
- БВ 2А типа характеризуется сниженным VWF, который не связывается с тромбоцитами, уменьшая способность тромбоцитов слипаться, образуя сгусток. Больные часто имеют легкое или умеренное слизисто-кожное кровотечение.
- БВ 2B типа характеризуется тромбоцитами, которые обладают повышенной способностью к слипанию, вызывая преждевременное слипание тромбоцитов в кровотоке, а не в месте повреждения кровеносного сосуда. Люди испытывают легкое или умеренное кровотечение из слизистой оболочки и имеют риск развития пониженного уровня тромбоцитов в крови (тромбоцитопения). Тромбоцитопения усугубляется стрессовыми ситуациями, такими как инфекция, операция или беременность.
- БВ 2M типа характеризуется сниженной активностью VWF и его неспособностью взаимодействовать с тромбоцитами. Эта форма обычно связана с легким или умеренным слизисто-кожным кровотечением. В некоторых случаях могут развиться более серьезные кровотечения.
- БВ 2N типа характеризуется неспособностью VWF транспортировать фактор VIII к месту повреждения и сниженным уровнем фактора VIII в крови. У людей развивается чрезмерное кровотечение после операции. Эта форма болезни Виллебранда может напоминать легкую форму классической гемофилии (гемофилия A).
- БВ 3 типа является наиболее тяжелой формой расстройства. Она составляет примерно 5% случаев. Пострадавшие люди имеют почти полное отсутствие VWF в крови. Больные могут испытывать сильное слизисто-кожное кровотечение, кровотечение в мышцах и суставах, повреждение суставов и развитие множественных гематом.
Для женщин с более тяжелыми формами БВ очень важен мониторинг серьезных, опасных для жизни, кровотечений в репродуктивном тракте и послеродового кровотечения.
Причины болезни Виллебранда
Большинство случаев болезни Виллебранда вызваны мутациями гена VWF. При БВ 1 типа и большинстве форм 2 типа мутация наследуется как аутосомно-доминантный признак. В некоторых случаях мутация происходит случайным образом без причины (спонтанно) без предшествующего семейного анамнеза (т.е. новой мутации). БВ 3 типа и некоторые случаи БВ 2 типа наследуются как аутосомно-рецессивный признак.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Аномальный ген может быть унаследован от любого из родителей или быть результатом новой мутации (изменения гена) у больного. Риск передачи ненормального гена от больного родителя к потомству составляет 50% при каждой беременности, независимо от пола ребенка.
Рецессивные генетические нарушения возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, передать оба дефектных гена и, следовательно, зачать больного ребенка, составляет 25% с каждой беременностью. Риск зачать ребенка-носителя как и родители, составляет 50% с каждой беременностью. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным для этой конкретной черты составляет 25%. Риск одинаков для мужчин и женщин.
Ученные определили, что ген VWF расположен на коротком плече (p) хромосомы 12 (12p13). Хромосомы присутствуют в ядре клеток и несут генетическую информацию каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин есть одна Х и одна Y-хромосома, а у женщин две Х-хромосомы. Каждая хромосома имеет короткое плечо, обозначенное «p», и длинное плечо, обозначенное «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 12p13» относится к полосе 13 на коротком плече хромосомы 12. Пронумерованные полосы указывают местоположение тысяч генов, присутствующих в каждой хромосоме.
Ген фактора VWF является единственным геном, идентифицированным как вызывающий болезнь Виллебранда (БВ). Ген VWF регулирует (кодирует) фактора фон Виллебранда. Мутации в этом гене приводят к низким уровням VWF фактора, который является дефектным и не функционирует должным образом. Как уже упоминалось выше, фактора фон Виллебранда выполняет две основные функции в организме. Он несет и защищает фактор VIII, предотвращая его расщепление (метаболизм) до места повреждения, и помогает тромбоцитам прилипать к кровеносным сосудам. Дефектный VWF фактор не может действовать как клей для удержания тромбоцитов в месте повреждения кровеносного сосуда. Следовательно, тромбоциты не прилипают к стенке кровеносного сосуда, и сгусток крови разрушается преждевременно. В некоторых случаях дефицит или дефект фактора фон Виллебранда приводят к низким уровням фактора VIII в крови, что приводит к аномальному образованию сгустков крови.
Несмотря на успехи, достигнутые в последние годы в отношении БВ, основные механизмы и генетика БВ 1 типа до конца не изучены. 1 тип БВ связан с переменной экспрессивностью и сниженной пенетрантностью среди членов семьи. Переменная экспрессивность относится к расстройствам, при которых степень тяжести от одного человека к другому может значительно различаться даже среди членов одной семьи. Пониженная пенетрантность означает, что у некоторых людей, унаследовавших дефектный ген для расстройства, не будут развиваться все симптомы.
Кроме того, только приблизительно 50-60% людей с БВ 1 типа имеют идентифицируемую мутацию гена фактора VWF. Даже в случаях, когда идентифицирована генная мутация, путаница все еще существует, потому что точная связь между мутациями гена фактора VWF, остаточными уровнями фактора VWF и общим риском кровотечения у людей с заболеванием 1 типа неясна.
Некоторая путаница вокруг болезни Виллебранда 1 типа проистекает из того факта, что люди в общей популяции имеют различные уровни фактора VWF в крови. В целом, при многих расстройствах количество остаточной активности белка часто коррелирует с тяжестью заболевания (например, незначительная или нулевая активность белка приводит к тяжелому заболеванию). Тем не менее, исследователи отметили, что многие люди в общей популяции имеют умеренно низкий уровень VWF, но у них не развиваются симптомы БВ. Это говорит о том, что дополнительные генетические и экологические факторы, вероятно, играют роль в развитии и тяжести БВ. Например, люди с группой крови 0 имеют более низкие уровни фактора VWF, чем люди с другими группами крови.
Из-за различий в уровнях VWF среди населения, трудно установить, что является «низким», а что «нормальным». Обычно считается, что нормальный диапазон VWF в крови составляет 50-200 МЕ/дл. Люди с очень низкими уровнями (например, <20 МЕ/дл), очень вероятно, имеют идентифицируемую мутацию гена фактора VWF, тяжелые симптомы кровотечения и положительную семейную историю расстройства. У этих людей может быть диагностирована БВ 1 типа. Тем не менее, люди с уровнями VWF между 30-50 МЕ/дл, что технически ниже нормы, представляют значительную проблему в отношении диагностики и определения.
Во многих случаях 1 тип БВ можно рассматривать как сложное генетическое заболевание, при котором множество факторов, как генетических, так и экологических, играют определенную роль в развитии заболевания. Генетические факторы, связанные с 1 типом БВ, могут включать факторы, не связанные с VWF. Необходимы дополнительные исследования, чтобы полностью понять сложные, лежащие в основе механизмы, которые в конечном итоге вызывают БВ типа 1, и помочь установить четкое, универсальное определение расстройства. До этого времени в медицинской литературе могут сохраняться разногласия относительно того, следует ли классифицировать лиц с умеренно низким уровнем фактора VWF и без симптомов как имеющих болезнь или имеющих фактор риска развития БВ или вообще не имеющих заболевание.
Затронутые группы населения
Болезнь Виллебранда (БВ) является распространенным наследственным нарушением свертываемости крови. Он поражает мужчин и женщин в равных количествах, хотя чаще диагностируется у женщин, потому что у женщин чаще встречаются симптомы (например, сильное менструальное кровотечение и кровотечение после родов). БВ может быть диагностирован в любом возрасте и у людей любой расы или этнической принадлежности.
По оценкам, заболевание поражает 1% населения. Тем не менее, распространенность симптоматического БВ оценивается в 23-110 на 1000000 в общей популяции. Распространенность БВ в медицинской литературе варьируется, поскольку для определения лиц, страдающих этим расстройством, используются разные критерии. Например, как указано выше, некоторые медицинские источники приравнивают низкий уровень VWF к наличию БВ, в то время как другие источники считают это фактором риска развития расстройства. Кроме того, некоторые случаи болезни Виллебранда остаются не диагностированными или неправильно диагностируются. Следовательно, трудно определить истинную частоту БВ среди населения в целом. Несмотря на это, все формы БВ типа 2 являются редкими расстройствами. БВ типа 3 встречается крайне редко, по оценкам, примерно у 1 из 250 000-1 000 000 человек в общей популяции.
Диагностика
Диагноз БВ основан на выявлении характерных симптомов (например, наличие слизисто-кожных кровотечений), детальной истории болезни пациента и семьи, тщательной клинической оценке и различных специализированных тестах. Такие тесты могут измерять количество VWF, насколько хорошо он функционирует, количество фактора VIII и способность крови к свертыванию.
Люди с тяжелыми случаями болезни Виллебранда могут быть диагностированы в младенческом возрасте. Легкие случаи БВ трудно идентифицировать и не могут быть диагностированы до совершеннолетия. Точный диагноз чрезвычайно важен для женщин, чтобы избежать ненужных и/или инвазивных методов лечения, таких как гистерэктомия, при ненормальном менструальном кровотоке.
Из-за разногласий и путаницы в медицинском сообществе относительно конкретного определения 1 типа БВ (см. раздел «Причины» выше), постановка диагноза может быть затруднена. Дифференцировать истинный 1 тип БВ от людей, которые имеют низкие уровни VWF, но не имеют расстройства, может быть сложной задачей.
Люди могут сдать стандартные анализы крови, включая общий анализ крови (ОАК), который может быть нормальным или демонстрировать микроцитарную анемию или низкий уровень тромбоцитов, особенно у людей с БВ типа 2B. Можно также использовать скрининговые коагулогические тесты, которые измеряют, сколько времени требуется, чтобы кровь свернулась. Два из этих тестов известны как активированное частичное тромбопластиновое время (АЧТВ) или время протромбина (ПТВ). АЧТВ может быть нормальным у людей с БВ или может быть продолжительным, если присутствует дефицит фактора VIII. ПТВ у людей с БВ обычно нормально.
Даже если вышеупомянутые скрининговые тесты являются нормальными, лица, подозреваемые на наличие БВ, должны пройти специальные анализы. Анализ — это тест, который может измерить активность определенных веществ в крови. Такие анализы включают тест на антиген VWF, который измеряет количество факторов VWF в крови; тест активности кофактора ристоцетина, который измеряет, насколько хорошо VWF работает, чтобы останавливать кровотечение; и тест свертывающей активности фактора VIII; который измеряет, насколько хорошо работает фактор VIII.
Если вышеупомянутые тесты положительны, больным необходимо будет пройти специализированное тестирование БВ, чтобы определить конкретный подтип истинного БВ. Такие тесты включают в себя мультимерный тест фактора фон Виллебранда, который исследует структуру VWF и может помочь определить конкретный тип присутствующего БВ и функциональные тесты тромбоцитов, которые определяют, насколько хорошо функционируют тромбоциты.
Диагноз болезни Виллебранда может быть подтвержден в некоторых случаях молекулярно-генетическим тестированием, которое может идентифицировать характерную мутацию гена фактора VWF, которая вызывает расстройство.
Лечение болезни Виллебранда
Излечивающих болезнь Виллебранда лекарств нет, но есть безопасные и эффективные методы управления всеми типами расстройства. Специфическое лечение БВ варьируется в зависимости от подтипа и тяжести расстройства. Незначительные кровотечения, такие как носовые кровотечения, небольшие ушибы и небольшие порезы, могут не требовать терапии. В легких случаях пациентам может потребоваться лечение только перед хирургическим вмешательством или стоматологической процедурой или после травмы или пореза. Лица с БВ должны получать быстрое лечение во время тяжелых кровотечений.
Лица с БВ могут получить направление в центр лечения гемофилии. Эти специализированные центры могут предоставлять комплексную помощь людям с гемофилией и связанными с ними расстройствами, включая разработку конкретных планов лечения, мониторинг и последующее наблюдение за больными людьми, а также современную медицинскую помощь. Лечение в центре гемофилии обеспечивает уход за отдельными лицами и членами их семей профессиональной командой медиков (врачей, медсестер, физиотерапевтов, социальных работников и генетических консультантов), имеющих опыт лечения пациентов с наследственными нарушениями свертываемости крови.
Пациентам с легким или умеренным БВ, вызванными травмой или спонтанными эпизодами кровотечений можно назначать препарат десмопрессина ацетат (Десмопрессин), который стимулирует высвобождение VWF, хранящегося в стенках кровеносных сосудов. Препарат представляет собой синтетический агент, являющийся производным природного гормона вазопрессина (антидиуретического гормона), также может повышать активность фактора VIII, позволяя крови сворачиваться должным образом и тем самым сокращая время кровотечения. Парентеральное применение десмопрессина также может быть использовано для контроля кровотечений (гемостаза) во время и после хирургических вмешательств у пациентов с легкими или умеренными заболеваниями. Некоторые женщины могут использовать препарат в начале менструального цикла. Десмопрессин можно вводить внутривенно, подкожно или доставлять через назальный спрей.
Десмопрессин является терапией первой линии для людей с болезнью Виллебранда 1 типа и некоторых людей с 2 типом. Терапия десмопрессином не показана для людей с большинством форм БВ 2 типа и тяжелом БВ. У людей с БВ типа 2B десмопрессин может потенциально вызвать снижение количества тромбоцитов, и его следует использовать только после того, как проведено тестирование перед терапевтическим использованием, чтобы определить реакцию больного на препарат. У большинства людей с БВ типа 2M реакция на десмопрессин слабая. Десмопрессин, как правило, также неэффективен при 3 типе БВ.
Лицам, которые не отвечают на лечение десмопрессином или имеют более тяжелые осложнения, может потребоваться лечение концентрированными формами VWF и фактора VIII. Это называется заместительной терапией, поскольку она заменяет белки, которые являются дефектными или отсутствуют в крови. Концентрированные формы VWF и фактора VIII создаются из плазмы тысяч различных доноров крови. Эти продукты подвергаются вирусной инактивации, которая убивает любые вирусы или подобные патогены, которые могут потенциально присутствовать в крови. Когда требуется лечение, заместительная терапия используется для людей с тяжелым БВ или людей с легкой или умеренной болезнью, которым противопоказана терапия десмопрессином. Заместительная терапия — это выбор лечения при БВ типа 2M и типа 3 и для некоторых людей с БВ типа 2B. Лицам с БВ 3 типа могут потребоваться повторные вливания концентратов фактора VWF/FVIII. Лица с 3 типом БВ часто получают концентраты VWF/FVIII в качестве профилактической терапии против развития мышечно-скелетного кровотечения и повреждения суставов.
Некоторых людей с поверхностным кровотечением можно лечить фибриновым клеем. Фибринный клей наносится непосредственно на место кровотечения. Фибрин является белком, который необходим для образования сгустка крови. Фибриновый клей изготовлен из различных факторов свертывания, полученных из донорской плазмы. Клей удерживает тромбоциты вместе, чтобы укрепить сгусток. Фибриновый клей часто используется для хирургических или стоматологических процедур у пострадавших людей.
Дополнительные методы лечения БВ включают гормональные контрацептивы, такие как противозачаточные таблетки, которые могут повысить уровень VWF в крови, помогая контролировать сильное кровотечение во время менструального цикла женщины. Препараты, известные как антифибринолитики, которые замедляют расщепление факторов свертывания крови, также могут быть использованы для лечения людей с БВ.
Люди с болезнью Виллебранда должны избегать лекарств, которые влияют на свертываемость крови, включая аспирин, лекарства, которые содержат аспирин, нестероидные противовоспалительные препараты (НПВП) и разжижающие кровь препараты, такие как варфарин и гепарин.
Источник: tvojajbolit.ru
