
Опущение миндалин мозжечка в большое затылочное отверстие или кагал спинного мозга называют дистопией. А иногда эта патология называется аномалией Киари. Как правило, такое заболевание не влечет за собой значительных расстройств или явных симптомов и не имеет причин для беспокойства больного. Часто эта патология проявляется после достижения 30 – 40-летнего возраста. Обнаруживается обычно при обследованиях по другим причинам. Потому об этом заболевании следует знать, чтобы оно не оказалось неожиданностью для больного.
Для того, чтобы понять, что такое низкое расположение миндалин мозжечка, необходимо четко знать клинические симптомы и как обнаруживается патология. А поскольку это состояние сопровождается частыми головными болями, нужно сначала установить их причину, а потом уже заниматься лечением. Такое заболевание выявляется при МРТ.
Причины появления
Дистопия мозжечка, как правило, является врожденной патологией. Она возникает при смещении какого-либо органа в эмбриональный период. Вторичной она бывает лишь при проведении частых пункций или при люмбальных травмах. Иных причин появления данного заболевания не выявлено.
Миндалины мозжечка очень сходны с теми, которые находятся в гортани. В нормальном положении они располагаются выше БЗО черепа. А отклонения в их развитии и положении могут повлечь за собой не только дистопию. Чаще всего встречается опущение миндалин мозжечка ниже уровня черепа.
До сих пор заболевание Киари является патологией, о причинах возникновения которого неврологи не пришли к одному мнению. Некоторые придерживаются мнения, что эта аномалия возникает при уменьшении габаритов ямки позади черепного выходного отверстия к спинному мозговому каналу. Это нередко приводит к таким последствиям в процессе роста тканей, которые расположены в коробке. Они выходят в затылочный выходной канал. Иные специалисты полагают, что заболевание начинает развиваться по причине увеличения объемов мозговых тканей головы. В этом случае мозг начинает выталкивать через заднюю черепную ямку в затылочное черепное отверстие мозжечок и его миндалины.
Вызывает такое прогрессирование выраженной аномалии и ее переход в «клинику», как гидроцефалия. При этом увеличиваются общие размеры мозга, особенно мозжечковые ткани. Патологию Киари вместе с недоразвитым связочным аппаратом мозга сопровождает дисплазия тканей кости. Потому любая черепно-мозговая травма нередко приводит к усилению снижения уровня нахождения миндалин и мозжечка.
Типы аномалий
Есть такие виды аномальных отклонений – дистопия и аномалия Киари.
В свою очередь, заболевание Киари разделяют на четыре различных типа:
- Тип I отличается расположением миндалин ниже уровня большого затылочного отверстия. Определяется такая патология, как правило, у подростков и у взрослых. Часто ее сопровождает скопление спинальной ликворной жидкости в центральном канале, где расположен спинной мозг пациента.
- Тип II характеризуется проявлением сразу после появления на свет. Кроме того, помимо миндалин, при втором типе патологии в отверстие затылочной части выходят червь мозжечка с частью продолговатого мозга и желудочек. Второй тип аномалии много чаще сопровождает гидромиелия, чем при патологии, описанной в первом случае. В большей части случаев такое патологическое отклонение связано с врожденными грыжами, образованными в различных отделах спинного мозга.
- III тип отличают опустившиеся сквозь отверстие не только миндалины, а и мозжечок вместе с тканями продолговатого мозга. Они размещаются в шейном и затылочном отделах.
- Тип IV представляет собой недоразвитие тканей мозжечка. Эта патология не сопровождается смещением их в каудальном направлении. Но при этом аномалию чаще всего сопровождает врожденная киста, располагающаяся в черепной ямке, и гидроцефалия.
II и III типы часто проявляются в сочетании с явлениями дисплазии нервной системы, например, с гетеротопией мозговых тканей коры, кистами отверстия и пр.
Симптомы
Самой распространенной среди аномалий является патология первого вида. При ней нередко возможно проявление ликворно-гипертензионного синдрома, а также церебелло-бульбарное и сирингомиелическое явления, нарушения работы нервных окончаний внутри черепа.
Ликворно-гипертензионный синдром – это боли в затылке и шейных мышцах, которые усиливаются во время чихания, кашля или напряжения шейных мышечных тканей. Часто боли сопровождаются рвотой, не связанной с приемами еды. Многие симптомы патологии проявляются в зависимости от того, в каком положении располагаются миндалины мозжечка относительно отверстия, которое находится в затылочной ямке черепа. Наблюдается также:
- повышенный тонус мышц шейного отдела;
- нарушения речевых функций;
- ухудшение работы органов зрения и слуха;
- отклонения при глотании;
- частые головокружения, сопровождаемые шумом в голове;
- ощущение вращения окружающей обстановки;
- непродолжительные обмороки;
- перепады давления при резких движениях;
- атрофия языка;
- сиплость голоса;
- нарушения дыхания и чувствительности разных частей тела;
- приступы онемения;
- нарушения в тазовых органах;
- ослабление мышц конечностей.
Аномалия II и III типов имеет похожие симптомы, заметные уже с первых мгновений после рождения малыша. Второй тип сопровождает шумное дыхание, а также неожиданные приступы остановки дыхания, нейропарез гортанных тканей. Наблюдаются также отклонения процесса глотания.
Признаки дистопии редко бывают очевидными. Но все же возможны неврологические проявления:
- приступы «стреляющих» болей в шейных мышцах при усилении напряжения или кашле;
- частые боли в области головы;
- приступы головокружений и обмороки.
Если опущение миндалин сильное, иногда наблюдается расширение канала, связывающего головной и спинной мозг, а также образовываются полости вокруг канала.
Диагностические методы
Основным современным методом диагностирования дистопии является МРТ. В этом случае ни КТ, ни рентгеновские исследования не дают полной картины патологии.
Для диагностирования синдрома Киари не подойдут никакие стандартные методы типа ЭЭГ, ЭхоЭГ или РЭГ, потому как они не позволяют точно поставить диагноз. Осмотр невролога тоже не определит аномалию. Все эти методы могут показать подозрение только на повышенное давление внутри черепной коробки. Рентгенографию черепа также не стоит делать, поскольку она показывает лишь аномалии костных тканей, которые могут сопровождать патологию. Потому до введения в диагностическую практику томографии диагностировать эту болезнь было проблематично. Современные же способы диагностирования позволяют точно определить патологию.
В случае качественной визуализации костных тканей вертебрального перехода такие методы, как МСКТ или КТ, не дают достаточно точной картины. Единственным достоверным способом диагностирования аномалии Киари на сегодня является только МРТ.
Поскольку проведение исследования этим методом требует неподвижности больного, маленьких детей погружают в искусственный сон с помощью лекарств. Проводится также МРТ спинного мозга. Она направлена на диагностику любых аномальных отклонений в работе нервной системы.
Лечение
Консервативные методы лечения возможны лишь при очень незначительных отклонениях. Все зависит от того, каково состояние больного на момент обращения к врачу. В этом случае лечение направлено на снятие болезненных симптомов нестероидными лекарствами или миорелаксантами. Необходима также коррекция режима.
Единственным эффективным методом лечения при обширных отклонениях является хирургическое вмешательство, которое заключается в расширении ямки черепа и пластики твердых мозговых тканей оболочки.
Показаниями для оперативного лечения являются:
- изнуряющие головные боли, которые не снимаются препаратами.
- нарастание мозговых проявлений, приводящее к инвалидности.
Если аномальное отклонение протекает без каких-то ощутимых признаков, лечения не требуется. В случаях возникновения болезненных ощущений в районе шеи и затылка проводится консервативная терапия, при которой используют анальгетики и асептические лекарственные вещества, а также миорелаксанты.
При сопровождении аномалии Киари нарушениями неврологических функций или когда консервативный курс терапии не дает результатов, назначается хирургическая операция.
Часто при лечебных курсах синдрома Киари используется метод краниовертебральной декомпрессии. Операция подразумевает расширение отверстия затылочной части за счет удаления части костной ткани, отсечения мозжечковых миндалин и части двух позвонков шеи. Благодаря этому, нормализуется оборот цереброспинальной жидкости в мозговых тканях в результате выполнения заплаты из аллотрансплантата или искусственного материала. Иногда синдром Киари лечат с помощью шунтирования, которое позволяет дренировать цереброспинальную жидкость из центрального канала. Посредством хирургической операции можно отвести цереброспинальную жидкость в сосуды органов груди или брюшины.
Прогноз
Аномалия Киари первого типа может протекать бессимптомно всю жизнь. И третий тип патологии почти всегда приводит к смертельному исходу, если не осуществить своевременное лечение. В случае появления неврологических признаков заболевания первого или последнего типов очень важно своевременно проведенное хирургическое лечение, потому как появившийся недостаток неврологических функций будет плохо восстанавливаться, даже если успешно провести манипуляции. По разным данным, результативность хирургической операции отмечается примерно в половине эпизодов.
Источник: glmozg.ru
Арнольда-Киари Аномалия I
Аномалия Арнольда-Киари I — опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. Может сочетаться с сирингомиелией, базилярной импрессией или инвагинацией, ассимиляцией атланта. Симптомы коррелируют от степени снижения. Методом выбора в диагностике является магнитно-резонансная томография (МРТ). Оперативное лечение, ламинэктомия (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки, применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев.
авторы Dr Henry Knipe and Dr Frank Gaillard et al.
Эпидемиология
Аномалия Арнольда-Киари I встречается чаще у женщин [2].
Клиническая картина
В отличие от пороков развития Киари II, III и IV, аномалия Арнольда Киари I часто остается бессимптомной.
Вероятность стать клинических проявлений пропорциональна степени опускания миндалин. Все пациенты с пролабированием миндалин больше 12 мм имеют какую-либо симптоматику, тогда как примерно в 30% пролабирование в диапазоне между 5 и 10 мм протекают бессимптомными [1].
Компрессия ствола мозга (продолговатого мозга) может вызывать сирингомиелию с соответствующими симптомами и клинической картиной (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др.
Сопутствующие заболевания
Сирингомиелия шейного отдела позвоночника встречается в
35% (варьирует от 20 до 56%), гидроцефалиия в 30% [1,3] случаев, в обоих случаях считается данные изменения развиваются в результате нарушения ликвородинамики, центральном канале и вокруг спинного мозга.
В
35% (23-45%) выявляются скелетные аномалии [1, 3]:
- платибазия / базилярная импрессия
- атланто-затылочная ассимиляция
- деформация Шпренгеля (Sprengel)
- синдром Клиппель-Фейля (Klippel-Feil)
Патология
Аномалия Арнольда Киари I характеризуется пролабированием миндалин мозжечка через большое затылочное отверстие, в основном в результате несоответствия между размерами мозжечка и задней черепной ямке. Аномалию Арнольда Киари I следует отличать от эктопии миндалин, которая протекает бессимптомно и является случайной находкой, при которой, миндалины выступают через затылочное отверстие не более чем на 3-5 мм [1-2].
Рентгенологические особенности
Патология выявляется путем измерения максимального расстояния на которое миндалины выступают ниже плоскости большого затылочного отверстия (условной линии между ophisthion и basion), значения используемые для постановки диагноза отличаются у разных авторов [2]:
- выше затылочного отверстия: норма
- 6 мм: аномалия Арнольда Киари 1
Некоторые авторы используют более простую градацию [1]:
- выше затылочного отверстия: норма
- 5 мм: аномалия Арнольда Киари 1
Положение миндалин мозжечка меняется с возрастом. У новорожденных миндалины расположены чуть ниже большого затылочного отверстия и спускаются ниже с ростом ребенка, достигая своей низшей точки в возрасте 5 — 15 лет. В дальнейшем они поднимаются на уровень большого затылочного отверстия [3]. Таким образом, снижение миндалин на 5 мм у ребенка будет скорее всего нормой, а взрослом возрасте данные изменения следует рассматривать с подозрением [3].
Современное объемное сканирование с высоким качеством сагиттальной реформации относительно хорошо визуализирует затылочное отверстие и миндалины, хотя отсутствие контрастности (по сравнению с МРТ) с трудом позволяет провести точную оценку. Чаще патология может быть заподозрена на аксиальных изображениях, когда мозговое вещество охватывает миндалины а спинномозговая жидкость представлена в малом количестве или отсутствует. Данное состояние называется, “переполнением“ затылочного отверстия.
МРТ исследование является методом выбора. Сагиттальные срезы наиболее оптимальны для оценки аномалии Арнольда Киари I. Осевые изображения так же дают картину «переполненого» затылочного отверстия.
Лечение и прогноз
Аномалии Арнольда Киари I можно разделить на три этапа (хотя мало данное деление практически не используется в повседневной практике):
- бессимптомная
- компрессия ствола мозга
- сирингомиелия
Оперативное лечение применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев. Оно состоит в ламинэктомии (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки.
История
Впервые была описана в 1891 году Хансом Киари, Австрийским патологоанатом (1851-1916).
Типы мальформации Арнольда-Киари, симптомы, диагностика
Ряд заболеваний головного мозга провоцирует морфологическую патологию, связанную с компрессией продолговатого мозга в большом затылочном отверстии. Мальформация Арнольда-Киари возникает при менингомелоцеле, сирингомиелии, гидроцефалии. Нозология приводит к летальному исходу. Если лечить нозологию вначале развития, предотвращается вклинение.
Патогенез мальформации Арнольда-Киари
Раньше патология считалась врожденной. Практические наблюдения, научные исследования позволили предположить приобретенную причину возникновения. Опасность нозологии заключается в блокаде движения цереброспинальной жидкости с последующим смертельным исходом.
Физиологически мозжечковые миндалины располагаются выше большого затылочного отверстия. Провоцирующие факторы обуславливают смещение до уровня первого или второго шейного позвонка.
Симптомы синдрома Арнольда-Киари (САК):
- Нарушения глотания;
- Дискоординация ходьбы;
- Затылочные боли.
Только у небольшого числа детей признаки патологии прослеживаются сразу после рождения. Малыш не может сообщить о затылочном болевом синдроме, но во время ходьбы прослеживается шаткая походка, невозможность выполнения пробы Реберга (пациент не может попасть пальцем в кончик носа). Назначение МРТ помогает провести диагностическую верификацию между синдромом Киари, Денди-Уокера, базилярной инвагинацией, платибазией, базиллярной импрессией, идиопатической сирингомиелией.
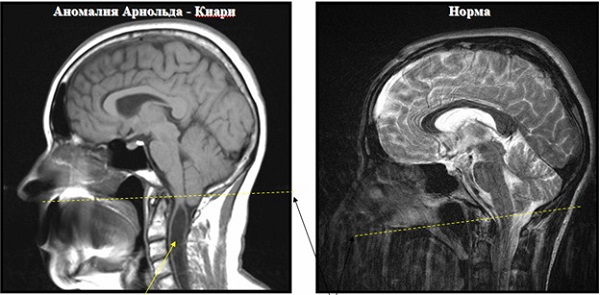
МРТ Арнольда-Киари
Виды мальформации Арнольда-Киари
- Опущение мозжечка при отсутствии мальформации – первый тип;
- Присутствие нейропозвоночных аномалий – второй тип;
- Наличие мозговых дефектов, затылочного энцефалоцеле – третий вид;
- Гипоплазия, аплазия мозжечка с присутствием намета;
- Внедрение мозжечка в большое затылочное отверстие, опущение структур.
При определении клинико-рентгенологической картины следует выделять морфологические формы заболевания:
- Задняя форма – характеризуется сдавлением задних корешков, спинномозговых нервов заднешейных отделов нервной системы, продолговатого мозга;
- Промежуточный тип обеспечивает компрессию верхнешейного и продолговатого отделов зубом второго шейного позвонка;
- Передняя форма – отклонение зуба аксиса кзади способствует формированию платибазии и базиллярной импрессии.
Морфологические изменения определяют особенности симптоматики патологии.
Клинические симптомы САК
При первой форма нозологии развивается гипермобильность суставов. Проявления возникают из-за разрастания соединительной ткани, возникающей при синдроме Марфана, Элерса-Данлоса. Состояние характеризуется нестабильностью краниоцервикальной области, последующим развитием мальформации.
Первый тип обуславливает также искривление позвоночного столба, сирингомиелия, гидроцефалия. По течению болезнь разделяется на хроническую и острую. У пятой части пациентов прослеживается быстрое нарастание симптомов.
Проявления определяются по большей части венозной обструкцией:
- Эпигастральный болевой синдром (закупорка мезентериальных сосудов);
- Желтушность кожных покровов (обтурация печеночных вен);
- Скопление жидкости внутри брюшной полости;
- Диарея.
В 95% случаев наблюдается хроническое течение синдрома Киари. Бессимптомное течение нозологии прослеживается несколько лет. Организм восстанавливает поврежденную микроциркуляцию посредством образования коллатералей. Опасность представляет кровоизлияние из варикозно-расширенных вен пищевода, кишечника.
Последствия синдрома Арнольда-Киари
Мальформация опасна тяжелыми осложнениями:
- Увеличение внутричерепного давления;
- Формирование печеночной недостаточности;
- Скопление жидкости внутри живота (асцит);
- Бактериальный перитонит;
- Портальная гипертензия;
- Печеночная карцинома;
- Гепаторенальный синдром;
- Внутрикишечные кровоизлияния.
Любое из описанных состояний является жизнеурожающим. Без экстренной медицинской помощи возникнет летальный исход.
Принципы диагностики САК
Определить первичные проявления нозологии позволяют общие, клинические методы, лабораторные, клинико-инструментальные способы.
Не сложно верифицировать острую форму САК по неврологическим проявлениям. Хроническое течение требует применения диагностических способов. С помощью МРТ головы удается определить опущение желудочков мозга, морфологический тип нозологии.
МР-ангиография – это исследование с контрастным веществом, позволяющее отслеживать обтурацию артерий, вен. Проводится после внутривенного введения специального, усиливающего парамагнитный эффект, препарата. Нативное сканирование обнаруживает опущение мозжечка больше, чем на 5 мм ниже большого затылочного отверстия.
Лучевая нейровизуализация позволяет провести дифференциальную диагностику между рядом схожих по клиническим симптомам патологическими состояниями:
- Начальные проявления аритмии;
- Венооклюзионная недостаточность;
- Тромбоз церебральных артерий.
Динамическое отслеживание состояния церебральных тканей позволяет предотвратить развитие сирингомелии из-за нарушения оттока спинномозговой жидкости. Сканирование позволяет спрогнозировать осложнения, провести своевременное лечение.
Для определения синдрома гипермобильности требуются томографы с поддержкой режима «Upright МРТ». Только такие устройства помогут верифицировать состояние.
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Диагностика мальформации Арнольда-Киари I типа по МРТ, КТ
Информация о работе и расписание
Госпитальная высококвалифицированная медицинская помощь
Услуги центра по восстановительной медицине
Современная диагностика – шанс предупредить болезнь
Он-лайн консультации для врачей по сложным практическим случаям
Трудоустройство в ФГАУ ЛРЦ
Стандарты и порядки оказания медицинской помощи
Проведение этической экспертизы клинических исследований, медицинских испытаний
Статьи и презентации
Мальформация Арнольда–Киари (Arnold-Chiari malformation) — мальформация цервико-медуллярного перехода, характеризуемая смещением миндалин мозжечка и в ряде случаев ствола и IVжелудочка ниже уровня большого затылочного отверстия.
Мальформация Киари I типа — смещение миндалин мозжечка вниз через большое затылочное отверстие к верхним отделам спинного мозга. Этот тип мальформации сопровождается гидромиелией и обычно проявляется в подростковом или взрослом возрастах. У подростков главные симптомы — нарушение сгибания и снижение силы в руках, утрата болевой и температурной чувствительности в верхней половине туловища и руках. Взрослые обычно жалуются на боль в шейно-затылочной области, возрастающую при кашле, а также боль в руках.
Мальформация Киари II типа характеризуется смещением червя мозжечка, миндалин, четвертого желудочка и продолговатого мозга (части ствола мозга) в большое затылочное отверстие. Данный тип, называемый также мальформацией Арнольд-Киари, гораздо чаще сопровождается гидромиелией, чем тип I и практически всегда связан с миеломенингоцеле. Миеломенингоцеле — это врожденное нарушение закрытия спинного мозга и позвоночника во время формирования плода. Симптомы этой мальформации очевидны и проявляются обычно сразу после рождения вместе с короткими эпизодами прекращения дыхания, сниженным глоточным рефлексом, непроизвольными и быстрыми движениями глазных яблок вниз, снижением силы в руках.
Мальформация Киари III типа заключается в смещении мозжечка и части ствола мозга с мозговыми оболочками в менингоцеле, расположенное в шейно-затылочной области.
Мальформации Арнольда–Киари II и III типов могут сопутствовать признаки дисплазии нервной системы: полимикрогирия, гетеротопия коры, гипоплазия подкорковых узлов, дисгенезия мозолистого тела, патология прозрачной перегородки, утолщение интерталамического соединения, beaking tectum (клювовидный tectum), часто отмечают наличие перегиба сильвиевого водопровода (55%), кисты отверстия Мажанди, гипоплазия серпа и намета мозжечка, hemivertebrae, низкое расположение каудального отдела спинного мозга на уровне LIV–V позвонков и ниже.
Этиология заболевания в настоящее время не ясна. Имеются данные, свидетельствующие о роли генетического фактора в этиологии этого синдрома. Эктопия миндалин мозжечка в затылочное отверстие была обнаружена у трех монозиготных близнецов. После первого описания мальформации Cleland в 1883 г. появилось несколько теорий. Теория, подтверждаемая исследованиями Misao Nishikawa и соавторов, заключается в том, что из-за парааксиальной дисплазии мезодермального листка или первичного повреждения структур соответствующего сомита формируется ненормально маленькая задняя черепная ямка, структуры заднего мозга, заполнив объем задней черепной ямки и продолжая расти, опускаются в затылочный канал. Сочетание Аномалии Киари II типа с менингомиелоцеле связано с тем, что степень парааксиальной дисплазии мезодермального листка при АК – II типа более выражена, чем при АК – I типа и отмечается не только на уровне формирования затылочной кости, но и по оси тела на уровне формирования ряда позвонков, что проявляется в spina bifida, а также в аномалиях ряда других костных структур и костной системы в целом.
Клинические проявления АК – I типа проявляются чаще всего в юношеском либо в зрелом возрасте. Эти проявления укладываются в такие неврологические синдромы, как церебеллобульбарный, ликворогипертензионный, сирингомиелический, синдромы повреждения черепных нервов. Ликворогипертензионный синдром проявляется головной болью, обычно субокципитальной, и болью в шее, усиливающейся при кашле, чихании и напряжении, застойными дисками зрительных нервов. Стволовые нарушения и расстройства функций черепных нервов проявляются в виде неустойчивых осциллопсий, тригеминальной дизестезии, снижения слуха, шума в ушах, головокружения, дисфагии, остановки дыхания во время сна, периодических обмороков (часто связанных с кашлем), нарушения контроля над ЧСС, АД при переходе из горизонтального положения в вертикальное, могут наблюдаться атрофия половины языка, паралич голосовых связок, стридор, спастический или комбинированный (больше в верхних конечностях) тетрапарез.
Мозжечковые расстройства — нистагм, дизартрия, атаксия. Симптомы, связанные с сирингомиелическими кистами — онемение, расстройство чувствительности, обычно по диссоциированному типу, а также нейроартропатия, нарушение функций тазовых органов, отсутствие брюшных рефлексов, мышечная гипотрофия. При этом ряд авторов отмечают несоответствие между локализацией, протяженностью кисты, кистозным индексом (отношение переднезаднего размера кисты к таковому размеру поперечника спинного мозга на уровне кисты), с одной стороны, и зоной гипестезии, распространенностью сегментарных расстройств поверхностной чувствительности, выраженностью мышечной гипотрофии и степенью пареза — с другой. АК II типа манифестирует у новорожденных и в раннем детском возрасте такими симптомами, как апноэ, стридор, билатеральный парез голосовых связок, нейрогенная дисфагия с назальной регургитацией, цианоз во время кормления, нистагм, гипотония, слабость, спастика в верхних конечностях, что может прогрессировать вплоть до тетраплегии. Мальформация Киари III типа встречается редко, клинические проявления ее такие же, как при АК II.
Стандартное рентгенологическое исследование может выявить лишь косвенные признаки мальформации АК, компьютерная томография также не дает четкой визуализации мягкотканных структур. Широкое внедрение МРТ в клиническую практику позволило решить большинство проблем, связанных с диагностикой аномалии Киари. Этому способствовали хорошая визуализация структур задней черепной ямки, краниовертебрального перехода, спинного мозга, отсутствие артефактов от костных структур.

МРТ пациента с АК — I типа и сигингомиелией.
Ориентиры задней черепной ямки, используемые в диагностике АК. d + e = длина ската; S = сфеноокципитальный синхондроз; d = длина основания сфеноидальной площадки от спинки турецкого седла и сфеноокципитального синхондроза до ската; e = длина между синхондрозом и basion; b = длина ствола мозга между плоскостью соединения среднего мозга и моста и медулло-цервикальным соединением; a = угол намета мозжечка по отношению к линии Твайнинга (Twining’s line); c = длина полушария мозжечка; DS = верхушка спинки турецкого седла; IOP = внутреннее возвышение затылочной кости; OP = opisthion; B = basion; TW = линия Твайнинга; McR (B to OP) = линия МакРи (McRae’s line). (заимствовано из Dimensions of the posterior fossa in patients symptomatic for Chiari I malformation but without cerebellar tonsillar descent, Raymond F Sekula и соавт.).
Федеральное государственное бюджетное учреждение
«Федеральный центр нейрохирургии»
Министерства здравоохранения Российской Федерации (г. Тюмень)
Мальформация Арнольда-Киари
Краткие сведения о нозологии
| Отделение: | Нейрохирургическое отделение №1 (детское) |
| Методы лечения (технологии): | Методы лечения мальформации Арнольда-Киари |
Аномалия Арнольда-Киари — это группа врожденных аномалий развития головного мозга, характеризующихся опущением миндалин мозжечка в большое затылочное отверстие со сдавлением продолговатого мозга и развитием соответствующих неврологических симптомов (нарушение чувствительности кожи конечностей, развитие параличей (отсутствие способности двигать конечностями), нарушение дыхания (неритмичность дыхания, редкие дыхательные движения)).
- Аномалия Арнольда-Киари 1го типа. Опущение части мозжечка (его выростов – миндалин) ниже плоскости большого затылочного отверстия со сдавлением ствола мозга.

- Аномалия Арнольда-Киари 2го типа. В затылочное отверстие, кроме миндалин мозжечка, спускается также продолговатый мозг и средний мозг (структуры, находящиеся чуть выше). Часто сопровождается незаращением дужек позвоночника, гидроцефалией (нарушением оттока ликвора из полостей головного мозга. Ликвор – жидкость, обеспечивающая питание и обмен веществ в головном мозге).
- Аномалия Арнольда-Киари 3го типа. При этой форме аномалии образуется грыжа мозга (энцефалоцеле), выходящая через дефект костей черепа. В эту грыжу попадает часть мозжечка, что проявляется нарушением равновесия, шаткостью походки, нистагмом (ритмичные колебательные движения глаз из стороны в сторону).
- Аномалия Арнольда-Киари 4го типа. Эта форма характеризуется врожденным недоразвитием мозжечка. Чаще всего заканчивается летальным исходом.
Клиническая картина:
Аномалия Арнольда-Киари может никак себя не проявлять и обнаружиться случайно, при проведении компьютерной или магнитно-резонансной томографии.
Однако возможны следующие ее проявления:
- боли в шейно-затылочной области, усиливающиеся при кашле и натуживании;
- головные боли;
- снижение остроты зрения;
- нистагм (колебательные движения глазных яблок);
- нарушение глотания (поперхивание при еде, питье);
- шаткость походки, неустойчивость, частые падения;
- расстройство координации движений: движения пациента нечеткие, некоординированные, размашистые;
- нарушение глубокой чувствительности (нарушается чувство восприятия собственного тела, например, человек с закрытыми глазами не ощущает, в каком положении у него лежат руки);
- снижение болевой и температурной чувствительности верхних конечностей;
- нарушения дыхания (вплоть до остановки) — в тяжелых случаях.
Диагностика:
- МРТ (магнитно-резонансная томография) головного мозга и шейного и грудного отделов спинного мозга позволяет достоверно выявить опущения структур головного мозга ниже уровня большого затылочного отверстия, а также определить наличие осложнений, связанных с нарушением циркуляции спинномозговой жидкости (гидроцефалия, сирингомиелия).
Показания к оперативному лечению:
- Опущение структур головного мозга ниже уровня большого затылочного отверстия;
- Неврологические нарушения, не купируемые консервативным путем;
- Признаки нарушений циркуляции спинномозговой жидкости(гидроцефалия, сирингомиелия).
Противопоказания к оперативному лечению:
- Воспалительный процесс любой локализации в стадии обострения, неполной ремиссии
- Анемия средней и тяжелой степени.
- Тяжелое общесоматическое состояние
- Обострение хронических заболеваний.
Окончательное решение о наличии или отсутствии показаний к оперативному лечению принимает врач-нейрохирург в рамках очной или заочной консультации.
Ожидаемый результат операции: Устранение сдавления структур мозжечка и продолговатого мозга, восстановление ликвороциркуляции.
Особенности послеоперационного периода:
Любое хирургическое вмешательство может быть с риском развития воспалительных, геморрагических осложнений.
Количество койко-дней: 6-10.
Необходимость в повторной явке: В послеоперационном периоде пациенту необходим динамически МСКТ/МРТ-контроль, с последующей консультацией нейрохирурга.
Источник: amt-market.ru
Аномалия Арнольда-Киари — что это
Справочно. Аномалия Арнольда-Киари — это нарушение развития, которое приводит к опущению мозжечковых миндалин ниже анатомического уровня и к ущемлению продолговатого мозга.
Нормой считается расположение миндалин мозжечка выше большого затылочного отверстия. В процессе мальформации они могут сместиться даже до уровня второго шейного позвонка. При таком смещении усиливается блокировка тока спинномозговой жидкости.
Не всегда удается распознать это заболевание сразу, а затем происходит его резкая манифестация. Проявление патологии происходит к 25 — 40 годам.
При обнаружении характерных для аномалии симптомов необходимо обратиться к врачу, в противном случае риск развития инфаркта спинного мозга значительно возрастает.
Поскольку речь идет об отклонении от нормального развития организма, заболевание часто называют мальформация Арнольда-Киари.
Особенности мальформации
Справочно. Задний отдел мозга перемещается к большому затылочному отверстию (БЗО), когда у черепной ямки оказываются слишком малые размеры. В итоге формируется порок, возникают нарушения циркуляции ликвора.
Кости черепа не позволяют БЗО менять свой диаметр, поэтому при любых смещениях мозговых структур ущемляются близлежащие ткани. Последствия такого явления могут носить для пациента фатальный характер.
Продолговатый мозг отвечает за работу сердечно-сосудистой и дыхательной системы организма. Его сдавливание приводит не только к неврологическому дефициту, но может давать более опасные последствия, вплоть до смерти больного.
Смещение правого и левого полушарий мозжечка останавливает движение ликвора, что провоцирует гидроцефалию. Водянка повышает риск осложнений тех расстройств, которые уже имеются у пациента.
Процент пациентов с врожденной формой аномалии Киари невелик.
Справочно. Последние данные говорят о приобретенном характере. Дистопия миндалин мозжечка обусловлена быстрым ростом тканей при медленно протекающих изменениях в строении черепа. В медицине также известна под названием эктопия миндалин мозжечка.
Синдром Арнольда-Киари без проявления симптоматики может случайно обнаружиться во время проведения МРТ.
Причины развития аномалии
Мнения медиков о причинах появления аномалии расходятся. Есть несколько теорий, объясняющих то, каким образом развивается порок.
Неврологи выделяют две патологии, приводящие к формированию мальформации Киари (Chiari malformation):
- Развитие плода в утробе матери может пойти с нарушением — черепная ямка окажется меньше анатомической нормы, отделы мозга приобретут обычные параметры.
- Размеры отделов увеличены, при этом параметры задней черепной ямки и БЗО отвечают нормам. Увеличивающийся мозг устремляется к отверстию.
Врожденная аномалия у ребенка развивается, если беременная женщина не контролирует прием лекарств, употребляет алкоголь, курит на ранних сроках беременности.
Кроме того, вирусные инфекции у будущей матери (краснуха, цитомегаловирус) могут пагубно сказаться на развитии плода.
Справочно. Возникновению заболевания могут предшествовать различные родовые травмы, гидроцефалия, сильные повреждения головы у взрослого человека.
Типы аномалии
Рассматриваются четыре типа. Классификация производится с ориентацией на определенные основания.
Существенными признаками оказываются следующие изменения: те, что произошли в головном мозге на структурном уровне, те, что говорят о недоразвитости черепной коробки.
Аномалия Арнольда-Киари 1 типа отличается сдвигом мозжечковых миндалин, сопровождается нарушением циркуляции ликвора.
Последний заполняет узкий канал спинного мозга, вызывая гидромиелию. Указанный тип аномалии носит благоприятный прогноз. Он часто диагностируется у подростковой и взрослой групп населения.
Аномалия Арнольда-Киари 2 типа проявляет себя у новорожденных детей. Здесь наблюдается еще большее смещение отделов. Помимо мальформации, у грудничков диагностируется спинномозговая грыжа, обнаруживается аномальное развитие позвоночного столба.
В области затылка происходит выпячивание мозгового вещества через мягкую оболочку, мозжечок оказывается там же. Такова картина аномалии Арнольда-Киари 3 типа.
Внимание. Аномалия Арнольда-Киари 4 типа дает о себе знать тем, что мозжечок новорожденного оказывается недоразвитым, не занимает должного анатомического положения. Такая патология делает младенца неприспособленным к жизни, летальный исход неизбежен.
Степени тяжести
Сколько живут с аномалией Арнольда-Киари 1-й степени? Такой вопрос часто задают люди, услышавшие свой диагноз. Такая степень тяжести является самой невысокой, клинические проявления могут не отмечаться.
Спровоцировать возникновение симптомов могут черепно-мозговые травмы и повреждения позвоночника в верхней его части. Также запустить процесс может развившаяся нейроинфекция.
Аномалии 2 и 3-й степени уже сопровождаются патологическими изменениями в нервной ткани. У больного часто обнаруживают:
- смещение мозгового вещества;
- кисты проводящих ликвор путей;
- недоразвитость некоторых извилин мозга;
- гипоплазию подкорковых узлов.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Возможные осложнения
Мальформация в некоторых случаях провоцирует достаточно опасные осложнения и может привести многих пациентов к инвалидности. Часто отмечаются увеличение ВЧД, дыхательные расстройства, апноэ, на фоне мальформации развиваются инфекционные заболевания легких и мочеполовой системы.
Справочно. Тяжело протекающая патология может стать причиной наступления комы, остановки в работе сердца, в итоге, быстрой смерти.
В запущенных случаях реанимация позволяет лишь поддерживать жизненно важные функции, сдавленный мозг восстановить практически невозможно.
Постановка диагноза
Осмотр невролога, сбор анамнеза представляют собой часть диагностики — они необходимы, но недостаточны.
Энцефалограмма, диагностика нарушения кровообращения в головном мозге и шейном отделе позвоночника могут косвенно показать наличие ВЧД.
С помощью рентгенографии, компьютерной томографии можно зафиксировать дефекты формирования черепной коробки. Но для определения состояния нервной ткани такие способы окажутся малоинформативными.
Справочно. МРТ сегодня считается единственно достоверным методом, позволяющим провести точную диагностику и своевременно распознать синдром Арнольда-Киари.
Процедура предполагает полное обездвиживание пациента, чего легко добиться от взрослого человека. Трудности возникают, когда пациентами являются маленькие дети. В этом случае необходимо применение общего наркоза.
Варианты лечения
После постановки диагноза лечение больного осуществляет нейрохирург или невролог. В исключительных случаях устранение аномалии возможно лишь путем проведения операции.
Если единственный симптом болезни — головная боль, то врачи ограничиваются медикаментозной терапией. Специалисты подбирают препараты:
- устраняющие воспаление (Найз, Ибупрофен, Диклофенак);
- анальгетики (Кеторол);
- спазмолитики (Мидокалм).
Справочно. Показанием к операции является сильное сдавливание отделов мозга, явно выраженные неврологические расстройства, если положительный эффект от приема лекарственных препаратов не наблюдается.
Оперативное вмешательство
Благодаря хирургическому вмешательству можно устранить чрезмерное давление на нервную ткань и привести в норму движение ликвора. Одной из проводимых операций является краниовертебральная декомпрессия, направленная на увеличение размеров задней черепной ямки.
Внимание. Декомпрессия относится к классу травматичных и рискованных операций. Согласно статистике, она приводит к осложнениям у каждого десятого пациента.
Риск летального исхода у прооперированных больных выше, чем у тех, кто операцию не переносил. Нейрохирурги стараются проводить такой тип вмешательства только в самых крайних случаях, когда налицо явные признаки сдавливания мозга.
Иной вариант устранения последствий мальформации предполагает шунтирование, которое способно обеспечить отвод ликвора из черепной коробки. Благодаря имплантации специальных трубок жидкость перетекает в грудную и брюшную полости, ВЧД снижается.
В наиболее острых случаях требуется немедленная госпитализация больного и проведение всего спектра терапевтических, профилактических и коррекционных процедур.
Прогноз выживаемости
Продолжительность жизни зависит от типа аномалии и степени ее тяжести. Первый тип позволяет сделать благоприятный прогноз, поскольку симптоматика либо вовсе отсутствует, либо возникает после получения травм головы.
Если никаких проявлений болезни нет, то продолжительность жизни больных такая же, как и у здоровых людей.
Для пациентов со вторым типом аномалии прогноз хуже, она переносится тяжелее.
Иногда борьба с очаговой неврологической симптоматикой не приносит плодов даже при активном лечении медикаментами. В таком случае требуется хирургическое вмешательство, чтобы впоследствии изменения по неврологической части были менее выраженными.
Справочно. Третий и четвертый типы мальформации являются самыми тяжелыми для пациентов, прогноз зачастую неблагоприятен.
Заболевание затрагивает важные структуры мозга, у больного отмечаются пороки внутренних органов. Часто функции ствола мозга страдают настолько, что нарушения оказываются несовместимыми с жизнью.
Источник: neuromed.online
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
-
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
-
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
- 3 тип — грубое смещение заднего мозга в позвоночный канал с высокой шейной или подзатылочной грыжей головного мозга и его оболочек и выраженным гипертензивно-гидроцефальным синдромом.
- 4 тип — гипоплазия (недоразвитие) мозжечка без смещения его вниз с эктопией продолговатого мозга.
- 0 тип. В 1998 году американский детский нейрохирург Берманс Искандер (В. J. Iskandar) с коллегами впервые ввел понятие «Киари 0» («Сhiari 0») в описании 5 пациентов, имеющих неврологические симптомы аномалии Арнольда — Киари с сирингомиелией и положением миндалин мозжечка на уровне большого затылочного отверстия. Этот тип так же называют «пограничным с Киари».
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Источник: www.katrenstyle.ru